Last update 👈
David Palecek, May 5, 2025
BGCs are found using GECCO tool on the assembled contigs (fasta files) which can be invoked from the dashboard at Galaxy. Another dashboard is used to retrieve and visualize the results.
Jupyter Notebooks for the dashboards can be found here
Running GECCO jobs on Galaxy¶
You will need an account on the galaxy earth-system for this NBs to work. Your Galaxy access data should be stored as environmental variables in the .env file at the root of the repository
GALAXY_EARTH_URL="https://earth-system.usegalaxy.eu/"
GALAXY_EARTH_KEY="..."BUG
For unknown reason the Binder version of the dashboard does not work.
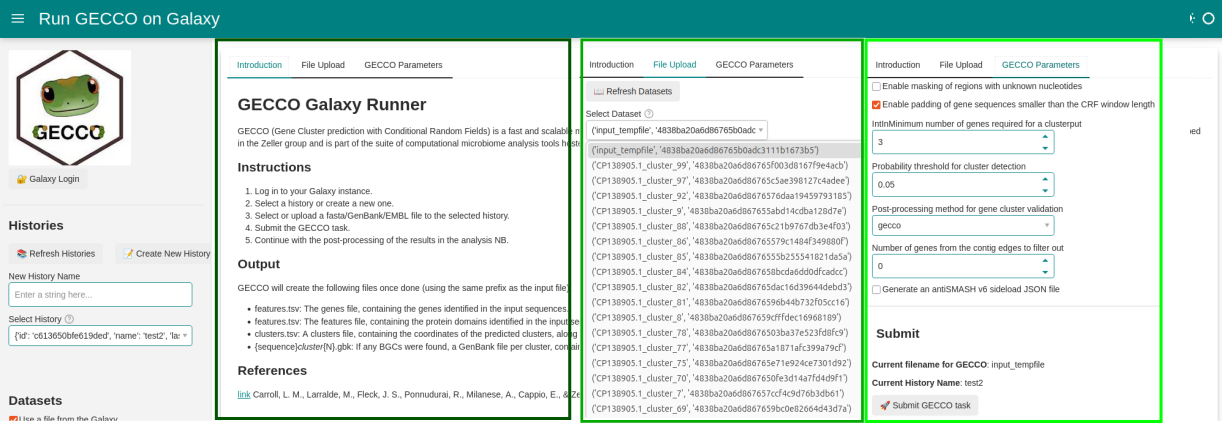
Dashboard illustrating submission of jobs to galaxy (GECCO tool):
- Upload and run workflow.
- Or start the workflow with existing data and in existing history on Galaxy.
- Monitor GECCO job.
Jupyter NB can be found here
Analyze GECCO results¶
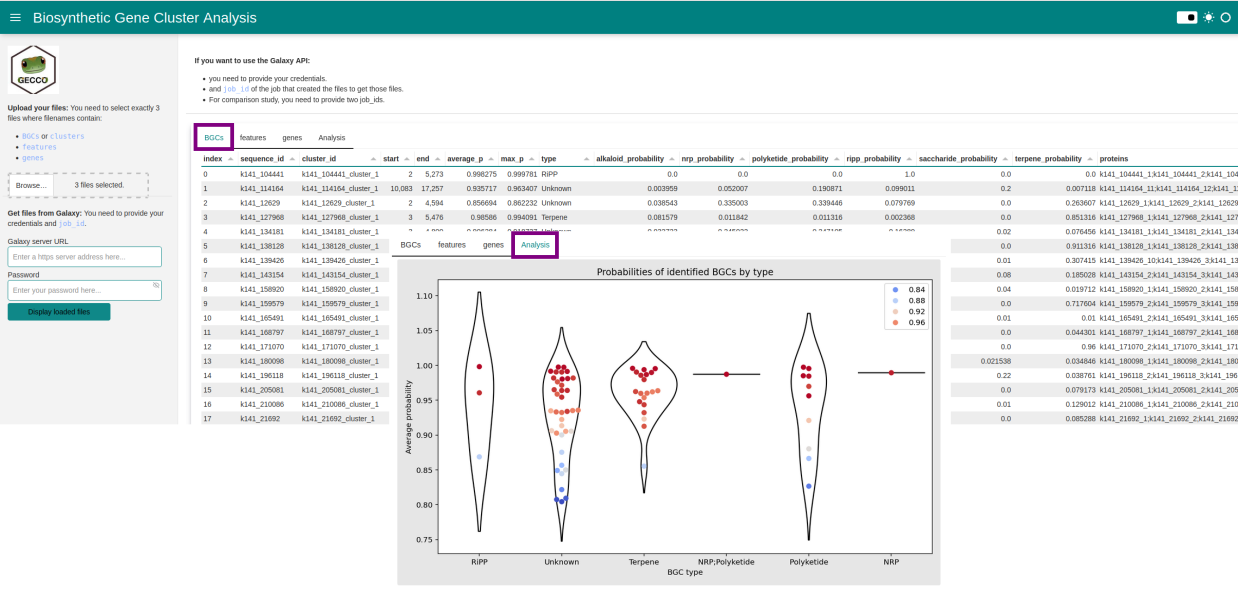
- Upload local data or query results of the GECCO from the Galaxy.
- Identifying Biosynthetic Gene Clusters (BGCs).
- Visualize BGCs.
Future directions¶
- Compare two samples in respect to each other (separate dashboard/NB)